66. H. V. Huynh, G. Frison* “Electronic structural trends in divalent carbon compounds“ J. Org. Chem., 2013, 78, 328–338.
DOI: 10.1021/jo302080c
This work aims to analyze and compare the intrinsic electronic densities in a series of neutral and anionic divalent carbon-donor derivatives. The σ-lone pair at the divalent carbon is the HOMO of these species. Structural factors have been identified that influence its energy, which is a measure of the σ-basicity. The π-electronic structure has been described as a function of the π-population. Our results show that no straightforward structural criteria correlate with the π-electronic distribution. However, the π-population, as well as the π-acidity and π-basicity, are related to the π-MOs. In all cases these π-MOs can be qualitatively obtained based on those of the protonated analogues by simply increasing the energy of the pπ orbital at the divalent carbon atom compared to normal sp2 carbon. Such an analysis allows a rationalization of the trends observed for the π-electronic structure of these ligands. Notably, this explains the values of the π-population at the divalent carbon center, which shows an increasing and continuous range from classical NHCs to mesoionic “carbenes”.

65. H. Sivaram, J. Tan, H. V. Huynh* “Syntheses, Characterizations, and a Preliminary Comparative Cytotoxicity Study of Gold(I) and Gold(III) Complexes Bearing Benzimidazole- and Pyrazole-Derived N-Heterocyclic Carbenes“ Organometallics 2012, 31, 5875−5883.
DOI: 10.1021/om300444c
WBS R-143-000-410-112
A series of Au(I) and Au(III) mono-, homobis-, and heterobis(carbene) complexes, [AuCl(FPyr)] (2), [Au(iPr2-bimy)2]PF6 (3), [Au(FPyr)2]PF6 (4), [Au(FPyr)(iPr2-bimy)]PF6 (5), [AuCl3(iPr2-bimy)] (6), [AuCl3(FPyr)] (7), [AuCl2(iPr2-bimy)2]PF6 (8), [AuCl2(FPyr)2]PF6 (9), and [AuCl2(FPyr)(iPr2-bimy)]PF6 (10), bearing the benzimidazole-derived iPr2-bimy (1,3-diisopropylbenzimidazolin-2-ylidene) and/or the pyrazole-derived FPyr (1,2,3,4,6,7,8,9-octahydropyridazino[1,2-a]indazolin-11-ylidene) N-heterocyclic carbene (NHC) ligands have been synthesized. Complexes 2–10 have been fully characterized using multinuclei NMR spectroscopy, ESI mass spectrometry, and elemental analysis. X-ray diffraction analyses have been performed on 2, 3, 5, 6, and 8. Together with the previously reported [AuCl(iPr2-bimy)] (1), the cytotoxic activities of all 10 complexes have been studied in vitro with the NCI-H1666 non-small cell lung cancer cell line. The cationic bis(carbene) complexes 3–5 and 8–10 show better cytotoxicity in comparison to cisplatin. In particular, the heterobis(carbene) complexes 5 and 10 have superior activity, with IC50 values of around 0.2 μM.

64. C. Bernhammer, H. V. Huynh* “Pyrazolin-5-ylidene Palladium(II) Complexes: Synthesis, Characterization, and Application in the Direct Arylation of Pentafluorobenzene” Organometallics 2012, 31, 5121–5130.
DOI: 10.1021/om300464b
WBS R-143-000-410-112
Ten palladium(II) complexes bearing a pyrazolin-5-ylidene ligand have been synthesized by oxidative addition and silver carbene transfer pathways. The weakly bound acetonitrile ligand in the initially obtained trans-[PdBr2(MeCN)(Pyry)] complex (6, Pyry = 1-phenyl-2,3-dimethylpyr-11 azolin-5-ylidene) could be replaced by other donor ligands, and additional NHC ligands were introduced either by silver carbene transfer reactions or via reaction with in situ generated free carbenes. Using our previously reported 13C NMR-based electronic parameter, the pyrazolin-5-ylidene ligand is estimated to be among the most strongly donating ligands on our scale so far. The complexes obtained were employed as catalysts for the direct arylation of pentafluorobenzene with moderate to good yields under optimized conditions.

63. S. Guo, H. V. Huynh* “Dipalladium Complexes with Triazolidin-Diylidene Bridges and Their Catalytic Activities” Organometallics 2012, 31, 4565−4573.
DOI: 10.1021/om3003625
WBS R-143-000-410-112
The 1,2,4-trimethyltriazolidin-3,5-diylidene (ditz) bridged dipalladium heterotetracarbene complex [PdBr
2(
iPr
2-bimy)]
2(
μ-ditz) (
3) (
iPr
2-bimy = 1,3-diisopropylbenzimidazolin-2-ylidene) was prepared by Ag–carbene transfer involving the 1,2,4-trimethyltriazolium dication (
C), Ag
2O, and the precursor complex (
iPr
2-bimyH)[PdBr
3(
iPr
2-bimy)] (
2). Bromido substitution of
3 with AgO
2CCH
3 and AgO
2CCF
3 afforded the carboxylato complexes [Pd(O
2CCH
3)
2(
iPr
2-bimy)]
2(
μ-ditz) (
4) and [Pd(O
2CCF
3)
2(
iPr
2-bimy)]
2(
μ-ditz) (
5). Multinuclei NMR spectroscopies and X-ray diffraction analyses showed that the all-trans isomers of
3,
4, and
5 are the predominant products in all three cases. In addition, the decomposition product of complex
4, trans-[Pd(O
2CCH
3)
2(
iPr
2-bimy)
2] (
trans–
6) was structurally determined by X-ray crystallography. A comparative catalytic study revealed the superiority of complexes
3,
4, and
5 over the previously reported mononuclear bis(benzimidazolin-2-ylidine) complexes without ditz bridges in the Mizoroki–Heck coupling reaction. Overall, complex
4 bearing acetato coligands showed the best catalytic performance.

62. J. C. Bernhammer, H. V. Huynh* “Correlation of spectroscopically determined ligand donor strength and nucleophilicity of substituted pyrazoles” Dalton Trans. 2012, 41, 8600–8608.
DOI: 10.1039/C2DT30526G
WBS R-143-000-410-112
The relative ligand donor strengths of 10 pyrazole-derived ligands has been determined with great accuracy, making use of the interdependence between the donor strength of the co-ligand and the
13C NMR chemical shift of the
iPr
2-bimy carbene signal in
trans-[PdBr
2(
iPr
2-bimy)L] complexes (
iPr
2-bimy = 1,3-diisopropylbenzimidazolin-2-ylidene; L = pyrazole-derived ligand). Even subtle variations in the substitution pattern of the pyrazole backbone up to three bonds away from the coordinating nitrogen could be detected reliably using this methodology. Alkylation experiments conducted on the pyrazoles using electrophiles of varied reactivity (ethyl bromide, ethyl iodide, and trimethyloxonium tetrafluoroborate) served as a benchmark to rank the pyrazoles in three groups of gradually increasing nucleophilicity, which correlated well with their determined donor strength.

61. D. Yuan, H. V. Huynh* “Sulfur-Functionalized N-Heterocyclic Carbene Complexes of Pd(II): Syntheses, Structures and Catalytic Activities” Molecules 2012, 17, 2491–2517.
DOI: 10.3390/molecules17032491
WBS R-143-000-407-112
N-heterocyclic carbenes (NHCs) can be easily modified by introducing functional groups at the nitrogen atoms, which leads to versatile coordination chemistry as well as diverse catalytic applications of the resulting complexes. This article summarizes our contributions to the field of NHCs bearing different types of sulfur functions, i.e., thioether, sulfoxide, thiophene, and thiolato. The experimental evidence for the truly hemilabile coordination behavior of a Pd(II) thioether-NHC complex has been reported as well. In addition, complexes bearing rigid CSC-pincer ligands have been synthesized and the reasons for pincer versus pseudo-pincer formation investigated. Incorporation of the electron-rich thiolato function resulted in the isolation of structurally diverse complexes. The catalytic activities of selected complexes have been tested in Suzuki-Miyaura, Mizoroki-Heck and hydroamination reactions.
60. H. Sivaram, R. Jothibasu, H. V. Huynh* “Gold Complexes of an Alicyclic Indazole-Derived N-Heterocyclic Carbene: Syntheses, Characterizations, and Ligand Disproportionation” Organometallics 2012, 31, 1195–1203.
DOI: 10.1021/om201268m
WBS R-143-000-410-112
A gold(I) NHC complex bearing a heteroalicyclic indazolin-3-ylidene ligand, [AuCl(Indy)] (1) (Indy = 6,7,8,9-tetrahydropyridazino[1,2-a]indazolin-3-ylidene), has been synthesized via the silver-carbene transfer method. Conversion of complex 1 to its heavier halido analogues [AuBr(Indy)] (2) and [AuI(Indy)] (3) was achieved by metathesis reactions involving LiBr and NaI in acetone, respectively. In contrast to 1 and 2, complex 3 undergoes ligand disproportionation/autoionization upon crystallization forming the solid complex salts [Au3I2(Indy)4][Au3I4(Indy)2] (3′) or [Au(Indy)2][AuI2] (3″) depending on the solvent used. This reversible process assisted by aurophilic interactions, and only occurring in the iodido complex 3, has been studied further by spectroscopic comparison with [Au(Indy)2]BF4 (4) and selective conversion of 3 to the gold(III) species [AuI3(Indy)] (5). All complexes 1–5 have been fully characterized using multinuclei NMR spectroscopies, ESI mass spectrometry and X-ray diffraction analysis.

59. D. Yuan, H. V. Huynh* “1,2,3-Triazolin-5-ylidenes: Synthesis of Hetero-bis(carbene) Pd(II) Complexes, Determination of Donor Strengths, and Catalysis” Organometallics 2012, 31, 405–412.
DOI: 10.1021/om2010029
WBS R-143-000-410-112
A series of hetero-bis(carbene) complexes trans-[PdBr2 (iPr2-bimy)(trz)] 1–4 (iPr2-bimy = 1,3-diisopropylbenzimidazolin-2-ylidene; trz = 1,2,3-triazolin-5-ylidene) bearing the constant iPr2-bimy and varying mesoionic 1,2,3-triazolin-5-ylidenes with different N-substituents has been synthesized as complex probes. Their 13C NMR spectroscopic evaluation shows that mesoionic 1,2,3-triazolin-5-ylidenes are in general stronger donors than classical NHCs, while weaker than some nonclassical NHCs such as pyrazolin-3-ylidenes and mesoionic imidazolin-4-ylidenes. More important and for the first time, this methodology proves useful in establishing substituent effects in the donating abilities of 1,2,3-triazolin-5-ylidenes on a finer level. In addition, the trifluoroacetato analogues [Pd(O2CCF3)2 (iPr2-bimy)(trz)] 5–7 have been synthesized through salt metathesis of 1, 2 and 4 with AgO2CCF3. The catalytic activities of complexes 1, 2 and 4–7 were examined in the direct arylation of pentafluorobenzene. Complexes bearing less donating trz ligands perform better in this catalysis, and trifluoroacetato complexes outperformed their bromido analogues.

58. D. Yuan, H. V. Huynh* “Synthesis and characterization of thiolato-functionalized N-heterocyclic carbene Pd(II) complexes with normal and mesoionic binding modes” Dalton Trans. 2011, 40, 11698–11703.
DOI: 10.1039/C1DT10789E
WBS R-143-000-407-112
(Cover article)
The thiolato-bridged dimeric Pd(II) NHC complex 1 has been synthesized from the reaction of thioester-functionalized imidazolium salt B and Pd(OAc)2. The isolation of its interesting constitutional isomer 2 bearing both classical C(2)-bound and mesoionic C(4)-bound ligands coordinating to two different metal centers in the same complex allowed for a direct comparison of these isomeric carbenes. Reactivity study of 1 with NaSCH(CH3)2 and NaBF4 afforded the tetranuclear compound 3 with a [Pd4S4] macrocycle. All complexes have been fully characterized by multinuclei NMR spectroscopies, ESI mass spectrometry and X-ray diffraction analysis.

57. H. V. Huynh*, R. Jothibasu “Syntheses and catalytic activities of Pd(II) dicarbene and hetero-dicarbene complexes” J. Organomet. Chem. 2011, 696, 3369–3375.
DOI: 10.1016/j.jorganchem.2011.07.018
WBS R-143-000-407-112
A series of palladium(II) complexes (1-6) bearing cis-chelating homo-dicarbene ligands with varying alkyl bridges (C1-C3) and N-heterocyclic backbones (imidazole and benzimidazole) have been synthesized by reaction of Pd(OAc)2 with the respective diazolium bromides (A•2HBr – F•2HBr) in DMSO. A comparative catalytic study employing aryl chlorides in the Mizoroki-Heck reaction revealed the superiority of methylene- and propylene-bridged dibenzimidazolin-2-ylidenes over their imidazole-derived analogues. Based on these results, two new propylene-bridged hetero-dicarbene complexes (7 and 8) were designed containing a mixed benzimidazole/imidazole derived NHC-donor set. Notably, both complexes outperformed their homo-dicarbene analogues, which may be due to the electronic asymmetry induced by hetero-dicarbene ligands. The molecular structures of complex 6 and 8 are also presented.
56. H. V. Huynh*, W. Sim, C. F. Chin “[2]Rotaxanes with Palladium(II)-NHC stoppers” Dalton Trans. 2011, 40, 11690–11692.
DOI: 10.1039/c1dt11472g
WBS R-143-000-407-112
Bridge cleavage reactions of the dimeric monocarbene complex [PdBr2(iPr2-bimy)]2 can be effectively used to end-cap pyridine containing pseudorotaxanes affording stable [2]rotaxanes.

55. D. Yuan, H. Tang, L. Xiao, H. V. Huynh* “CSC-pincer versus pseudo-pincer complexes of palladium(II): a comparative study on complexation and catalytic activities of NHC complexes” Dalton Trans. 2011, 40, 8788–8795.
DOI: 10.1039/C1DT10269A
WBS R-143-000-407-112
(Cover article and invited contribution for a special issue “Pincers and other hemilabile ligands” and “Hot article”)
Three thioether bridged diimidazolium dibromides with different steric and electronic properties have been synthesized as precursors to carbene-based CSC pincer ligands. Palladation afforded CSC Pd(II) pincer complexes for bulky and electron rich ligand systems, whereas the least donating ligand led to the formation of a pseudo-pincer complex. All complexes have been fully characterized by multinuclei NMR spectroscopies, ESI mass spectrometry and X-ray diffraction analysis. The catalytic activities of pincer versus pseudo-pincer complexes have been compared in the intermolecular hydroamination of alkynes with anilines as well.

54. L. Xue, L. Shi, Y. Han, C. Xia, H. V. Huynh*, F. Li* “Pd-carbene catalyzed carbonylation reactions of aryl iodides” Dalton Trans. 2011, 40, 7632–7638.
DOI: 10.1039/C1DT10433K
WBS R-143-000-407-112
A series of carbene complexes [PdBr2(iPr2-bimy)L] (C2-C13) with different types of co-ligands (L) have been tested for their catalytic activities in the carbonylative annulation of 2-iodophenol with phenylacetylene in DMF to afford the respective flavone 2a. Complex C12 with an N-phenylimidazole co-ligand showed the best activity and also afforded high yields when the substrate scope was extended to other aryl or pyridyl acetylenes. In addition, catalyst C12 was also efficient in the carbonylative annulation of 2-iodoaniline with acid chlorides giving the desirable 2-substituted 4H-3,1-benzoxazin-4-ones (4) in good yields. Additionally, this Pd-NHC complex also proved to be a very efficient catalyst for the hydroxycarbonylation of iodobenzene derivatives at low catalyst loading and under low CO pressure. These results demonstrate the versatility and efficiency of this phosphine-free Pd(II)-NHC complex in different types of carbonylations of aryl iodides under mild conditions.

53. H. V. Huynh*, Invited Book Review “Functionalised N-Heterocyclic Carbene Complexes” by Olaf Kühl, Wiley, 2010, 364 pp. (hardback) ISBN 978-0-470-71215-3″ App. Organomet. Chem., 2011, 25, 565.
DOI: 10.1002/aoc.1768
52. Y. Han, D. Yuan, Q. Teng, H. V. Huynh* “Reactivity Differences of Pd(II) Dimers bearing Heterocyclic Carbenes with two, one or no α-Nitrogen Atoms toward Isocyanides” Organometallics 2011, 30, 1224–1230.
DOI: 10.1021/om101169x
WBS R-143-000-410-112
Pd(II) dimers [PdI2(rNHC)]2 (1a/b) of pyrazole-based remote N-heterocyclic carbenes (rNHCs) have been synthesized through oxidative addition of 4-iodopyrazolium iodides (A/B) to [Pd2(dba)3]. Reaction of 1a with aromatic (CN-Xyl) or aliphatic (CN-Cy) isocyanides led to the template-assisted formation of novel Pd(II) dimers 8/9 bearing betainic C-imino ligands via isocyanide insertion into Pd-CrNHC bonds and subsequent dimerization. In contrast, both isocyanides reacted with dimers [PdI2(Me2-indy)]2 (2) and [PdI2(Me2-bimy)]2 (3) bearing indazolin-3-ylidenes and benzimidazolin-2-ylidenes under formation of mononuclear mixed carbene/isocyanide complexes 4-7. Notably, only dimer 8 underwent further bridge-cleavage with excess isocyanide yielding the mixed C-imino/CN-Xyl complex 10, while dimer 9 remained intact. These results highlight the uniquely different reactivity of complexes with carbenes with no α-nitrogen versus those with one or two α-nitrogen atoms as a result of their decreasing donor abilities.

51. Y. Han, H. V. Huynh* “Pyrazolin-4-ylidenes: a new class of intriguing ligands” Dalton Trans. 2011, 40, 2141–2147.
DOI: 10.1039/c0dt01037e
WBS R-143-000-410-112
(invited contribution for a special issue “New Talent: Asia”)
The immense success of N-heterocyclic carbenes in recent years has initiated the search for even stronger ligands leading to the discovery of abnormal and remote NHCs. This article reflects our particular interest in the coordination chemistry of pyrazolin-4-ylidenes as a contribution to this field. A modular approach to 4-iodopyrazolium salts with different substitution patterns is described, which upon oxidative addition to Pd0 gave rise to a library of new Pd(II) pyrazolin-4-ylidene complexes. A preliminary study showed that selected complexes are active precatalysts in Suzuki–Miyaura and Mizoroki–Heck coupling reactions. The 3,5-substituents of pyrazolin-4-ylidenes are found to have significant effects on complexation and catalytic activities. Finally, the nature of this new class of ligands is discussed, and a future direction for further explorations in this exciting field is envisioned.

50. A. D. Yeung, P. S. Ng, H. V. Huynh* “Co-ligand Effects in the Catalytic Activity of Pd(II)-NHC Complexes” J. Organomet. Chem. 2011, 696, 112–117.
DOI: 10.1016/j.jorganchem.2010.08.017
WBS R-143-000-407-112
(invited contribution for a special issue on Catalytic addition of E-H bonds to non-activated carbon-carbon multiple bonds)
Three cis-chelating di-N-heterocyclic carbene palladium(II) complexes [PdX2(diNHC)] (X = I, 1; X = SCN, 2; X = CF3CO2, 3) bearing different anionic co-ligands were synthesized and fully characterized. A comparison of their catalytic activities in the Mizoroki-Heck reaction and conjugate addition of arylboronic acids to cyclic enones revealed increasing efficiency in the order SCN– < I– < CF3CO2–. The di(trifluoroacetato) complex 3 showed the best activity in both transformations highlighting the importance of co-ligands effects in catalysis. In addition, the molecular structure of an unusual poly-heteronuclear complex salt 4 is reported, which has been isolated as a byproduct in the synthesis of complex 3.

49. D. Yuan, H. V. Huynh* “Dinuclear and Tetranuclear Palladium(II) Complexes of a Thiolato-Functionalized, Benzannulated N-Heterocyclic Carbene Ligand and Their Activities toward Suzuki-Miyaura Coupling” Organometallics 2010, 29, 6020–6027.
DOI: 10.1021/om1008023
The thiolato-bridged dimeric Pd(II) benzimidazolin-2-ylidene complex 1, with a [Pd2S2] core, was conveniently prepared by the reaction of the thiol-functionalized benzimidazolium salt C and Pd(OAc)2. More straightforwardly, 1 can also be synthesized by direct treatment of the thioester-functionalized benzimidazolium salt B with Pd(OAc)2 in wet DMSO under in situ hydrolysis of the thioester function. A subsequent salt metathesis reaction of 1 with AgO2CCF3 afforded the mixed dicarboxylato/NHC analogue 2 in quantitative yield, leaving the sulfur bridges of the [Pd2S2] core unaffected despite the use of the soft Ag(I) ions. Treatment of 1 with Me3OBF4 resulted in an unexpected bromido abstraction of 1 leading to an unusual rearrangement/dimerization reaction to give the tetranuclear NHC complex 3, which features a [Pd2S2] macrocylic square with sulfur corners. These reactions demonstrate the structural diversity of the thiolato-functionalized N-heterocyclic carbene complexes and may offer access to metallo-NHC-based supramolecular architectures. A comparative catalytic study revealed the superiority of NHC/thiolato complex 2 over complexes 1 and 3 in aqueous Suzuki-Miyaura couplings at very low catalyst loading.

48. R. Jothibasu, K.-W. Huang, H. V. Huynh* “Synthesis of cis– and trans-Diisothiocyanato-Bis(NHC) Complexes of Nickel(II) and Applications in the Kumada-Corriu Reaction” Organometallics 2010, 29, 3746–3752.
DOI: 10.1021/om100241v
Metathetical reaction of AgSCN with a series of trans-dihalido-bis(carbene) nickel(II) complexes in CH3CN readily afforded the novel diisothiocyanato-bis(carbene) complexes [Ni(NCS)2(NHC)2] (trans–2a, NHC = 1,3-diisopropylbenzimidazolin-2-ylidene; trans–2b, NHC = 1,3-diisobutylbenzimidazolin-2-ylidene; trans–2c, NHC = 1,3-dibenzylbenzimidazolin-2-ylidene; cis–2d, NHC = 1,3-di(2-propenyl)benzimidazolin-2-ylidene; cis–2e, NHC = 1-propyl-3-methylbenzimidazolin-2-ylidene) as greenish yellow powders in moderate to good yields. While dihalido-bis(carbene) Ni(II) complexes exclusively form trans-complexes, a trans–cis isomerization occurs upon halido-isothiocyanato exchange with complexes bearing less bulky carbene ligands, i.e. cis–2d/e. DFT calculations indicated that this isomerization can be attributed to a reduced energy difference between trans and cis isomers of diisothiocyanato complexes. All complexes have been characterized by multinuclear NMR spectroscopy, ESI mass spectrometry and X-ray diffraction analysis. A catalytic study revealed that cis-complexes generally exhibit greater activities in the Kumada-Corriu coupling reaction.

47. H. V. Huynh*, Y. X. Chew “Synthesis, structural characterization and catalytic activity of a palladium(II) complex bearing a new ditopic thiophene-N-heterocyclic carbene ligand” Inorg. Chim. Acta 2010, 363, 1979–1983.
DOI: 10.1016/j.ica.2009.02.035
(invited contribution for a special issue on Metals in Organic Chemistry)
A new thiophene-functionalized benzimidazolium salt (2) has been prepared by reacting N-methylbenzimidazole with 2-bromomethylthiophene (1), which in turn was obtained by bromination of 2 thiophenemethanol with PBr3. Subsequent reaction of salt 2 with Pd(OAc)2 afforded the cis configured bis(carbene) Pd(II) complex (cis–3), which in solution exists as an inseparable mixture of cis–anti and cis–syn-rotamers in a 3.5:1 ratio. All new compounds have been fully characterized by spectroscopic and spectrometric methods. A preliminary catalytic study shows that cis–3 is highly active in the Suzuki-Miyaura coupling of aryl bromides with phenylboronic acid in/on water as environmentally benign reaction media.

46. R. Jothibasu, H. V. Huynh* “Versatile coordination chemistry of indazole-derived carbenes” Chem. Commun. 2010, 46, 2986–2988.
DOI: 10.1039/b925977E
The versatile coordination chemistry of strongly donating indazolin-3-ylidene ligands as new members of the NHC family is demonstrated on 12 new PdII, AuI and RhI complexes, which were fully characterized by multinuclear NMR spectroscopies, ESI mass spectrometry and X-ray diffraction studies.

45. H. V. Huynh*, C. H. Yeo, Y. X. Chew “Syntheses, Structures, and Catalytic Activities of Hemilabile Thioether-Functionalized NHC Complexes” Organometallics 2010, 29, 1479–1486.
DOI: 10.1021/om9010966
Four imidazolium (5a/b) and benzimidazolium (6a/b) salts with hemilabile alkyl-aryl thioether functions have been prepared via a straightforward and modular pathway in order to compare their reactivities toward palladation. Reaction of 5a/b with Pd(OAc)2 gave complex product mixtures, whereas 6a/b afforded the desired bis(benzimidazolin-2-ylidene) complexes 8a/b in good yields. The difference in reactivities of benzimidazole versus imidazole derivatives was attributed to the presence of additional acidic protons at C4/5 positions of the imidazolium ring, leading to competing and unselective deprotonation reactions. The milder Ag-NHC transfer reaction, on the other hand, provided either mono- or bis(imidazolin-2-ylidene) complexes (9 or 7a/b) in good yields depending on the ligand:metal ratio. The interesting hemilability in monocarbene complex 9 was investigated by spectroscopy and thioether displacement reaction with PPh3, yielding the mixed NHC-PPh3 complex 10 in high yields. An initial comparative catalytic study also reveals that the mixed-donor complex 10 exhibits the highest activity among the complexes tested.

44. Y. Han, L. J. Lee, H. V. Huynh* “Pyrazole-Derived Remote Dicarbenes: Versatile Ligands for Di- and Tetranuclear Complexes” Chem. Eur. J. 2010, 16, 771-773.
DOI: 10.1002/chem.200902737
WBS R-143-000-410-112
Influence your neighbor: The first examples of pyrazole-based dicarbene complexes are described (for an example see figure). Remote changes in the ligand topology four bonds away from the carbon donor have substantial influences on the nuclearity of the resulting complexes.

43. S. K. Yen, D. J. Young, H. V. Huynh, L. L. Koh, T. S. A. Hor* “Unexpected coordination difference in geometric-isomerism between N,S– and N,N-heterocyclic carbenes in cyclometallated platinum(II)” Chem. Commun. 2009, 6831-6833.
DOI: 10.1039/b914036k
The reaction of [PtII(2-phenylpyridine)(acac)] and benzothiazolium bromide yields the N,S-heterocyclic carbene ligand trans to pyridyl while, surprisingly, a very similar N,N-heterocyclic carbene coordinates predominantly trans to the cyclometallated carbon.

42. H. V. Huynh*, Y. Han, R. Jothibasu, J. A. Yang “13C NMR Spectroscopic Determination of Ligand Donor Strengths using N-Heterocyclic Carbene Complexes of Palladium(II)” Organometallics 2009, 28, 5395–5404.
DOI: 10.1021/om900667d
The electronic parameters of 25 Werner-type and organometallic ligands have been experimentally determined and ranked on an unprecedented unified 13C NMR scale using safe and easily obtainable complexes of the type trans-[PdBr2(iPr2-bimy)L]n- (iPr2-bimy = 1,3-diisopropylbenzimidazolin-2-ylidene; L = ligand in question) as spectroscopic probes. The methodology is based on the sensitivity of the constant iPr2-bimy carbene signal to the donor strengths of the varying co-ligands, which even allows detection of backbone and substituent effects more accurately than previous carbonyl-based systems. For the evaluation of N-heterocyclic carbenes (NHCs), a one-pot approach to novel hetero-bis(carbene) complexes bearing two different NHCs is introduced. Furthermore, the first complex of a strongly donating indazolin-3-ylidene ligand is presented. The molecular structures of ten complex-probes have been characterized by single-crystal X-ray diffraction analyses.

41. S. K. Yen, L. L. Koh, H. V. Huynh, T. S. A. Hor* “Benzothiazolin-2-ylidene and Azole Mixed-Ligand Complexes of Palladium” Eur. J. Inorg. Chem. 2009, 4288–4297.
DOI: 10.1002/ejic.200900447
A series of mixed-ligand, mononuclear complexes trans-[PdX2(NSHC)(azole)] [NSHC = N,S-heterocyclic carbene; azole = imidazole, 1-(2,4,6-trimethylphenyl)-1H-imidazole, benzimidazole, benzothiazole and benzoxazole] have been prepared and characterised by X-ray single-crystal diffraction. These complexes catalyse the sp2–sp3 cross-coupling of fluoroaryl halides with arylboronic acid to give diarylmethanes with good yields and high turnovers.

40. H. V. Huynh*, D. Yuan, Y. Han “Syntheses and catalytic activities of pseudo-pincer and CSC pincer-type Pd(II) complexes derived from benzannulated N-heterocyclic carbenes” Dalton Trans. 2009, 7262-7268.
DOI: 10.1039/b907887h
(invited contribution for a special issue on NHC chemistry)
Reaction of thioether- and sulfoxide-functionalized dibenzimidazolium dibromides B⋅ 2HBr and C⋅ 2HBr with Pd(OAc)2 afforded two new pseudo-pincer complexes cis-[PdBr2(B–κ2C)] (1) and cis-[PdBr2(C–κ2C)] (2), which contain pendant sulfur-functions. On the other hand, palladation of the dinitrate analogue B⋅ 2HNO3 in the presence of 1 equiv of KBr yields the first NHC-based CSC pincer complex [PdBr(B–κ3CSC)]NO3 (3). All three complexes have been fully characterized by multinuclei NMR spectroscopies, ESI mass spectrometry and X-ray diffraction analysis. Their catalytic activities in the Mizoroki-Heck reaction are reported as well.

39. Swee Kuan Yen, Lip Lin Koh, Han Vinh Huynh, T. S. Andy Hor* “Structures and Suzuki-Coupling of N-Heterocyclic Carbene Complexes of PdII with Coordinated Solvent and PPh3” Aust. J. Chem. 2009, 62, 1047–1053.
DOI: 10.1071/CH09196
(invited contribution for the special issue “UQ-NUS Symposium”)
A series of mononuclear N,N-heterocyclic carbene (NNHC) complexes of PdII with mixed ligands of 1,3-dibenzylbenzimidazoly-2-ylidene and solvate (dimethyl sulfoxide, CH3CN, N,N-dimethylformamide, and pyridine) or PPh3 were prepared and characterized by X-ray single-crystal diffraction analysis. They are more active in the Suzuki–Miyaura coupling of selected aryl bromides than their N,S-heterocyclic carbene (NSHC) analogues.
38. H. V. Huynh*, H. X. Seow “Synthesis and Structural Characterization of Palladium Dicarbene Complexes bearing labile co-Ligands” Aust. J. Chem. 2009, 62, 983-987.
DOI: 10.1071/CH09143
(invited contribution for the special issue “UQ-NUS Symposium”)
Dicarbene complexes [Pd(OAc)2(diNHC)] (2), [Pd(O2CCF3)2(diNHC)] (3) and [Pd(CNCH3)2(diNHC)](SO3CF3)2 (4) bearing labile acetato, fluoroacetato and acetonitrile co-ligands have been synthesized via metathesis reaction of the respective precursor [PdBr2(diNHC)] (1) with Ag-salts. All complexes are stable toward air and moisture and have been fully characterized by spectroscopic and spectrometric methods. Notably and unlike diphosphine analogues, they resist ligand disproportionation in solution. Their molecular structures have also been determined by single crystal X-ray diffraction. A preliminary catalytic study showed low activity in the hydroamination reaction, but revealed an interesting co-ligand influence.

37. Y. Han, H. V. Huynh* “Pd(II) Pyrazolin-4-ylidenes: Substituent-Effects on the Formation and Catalytic Activity of Pyrazole-based Remote NHC Complexes” Organometallics 2009, 28, 2778-2786.
DOI: 10.1021/om8010849
Three 4-iodopyrazolium salts with 3,5-dimethyl (3a), 3,5-diphenyl (3b) and 3,5-diisopropyl (3c) substituents, respectively, were synthesized using a modular approach. The oxidative addition of 3a-c to Pd2(dba)3/PPh3 afforded products (trans–4a, cis-/trans–4b and 4c) of different geometries or connectivities indicating a dramatic substituent effect on the formation of pyrazolin-4-ylidene complexes. In addition, the reactions of 3a-c with Pd2(dba)3 in the presence of pyridine yielded new mixed pyrazolin-4-ylidene/pyridine complexes (5a-c). All complexes have been fully characterized by multi nuclei NMR spectroscopies, ESI mass spectrometry and X-ray diffraction analyses. Furthermore, an initial catalytic study in Suzuki-Miyaura and Mizoroki-Heck cross-coupling reactions also reveals a significant substituent effect on catalytic activities.

36. R. Jothibasu, H. V. Huynh* “Mixed Azido-N-Heterocyclic Carbene Complexes of Ni(II) as Template for New Organometallics bearing Carbodiimido, Tetrazolato and Abnormal Tetrazolin-5-ylidene Ligands” Organometallics 2009, 28, 2505–2511.
DOI: 10.1021/om900140e
Salt metathesis reaction of the dihalo-bis(carbene) complex trans-[NiBr2(NHC)2] (1, NHC = 1,3-diisopropylbenzimidazolin-2-ylidene) with NaN3 in DMF at elevated temperature afforded the diazido-bis(carbene) complex trans-[Ni(N3)2(NHC)2] (2) as a red crystalline solid in a yield of 78%. Complex 2 served as metal-template for the 1,3-dipolar cycloaddition of 2,6-dimethylphenylisocyanide (CN-Xyl) to the azido ligands to yield a mixed tetrazolato-carbodiimido complex trans-[Ni(CN4-Xyl)(NCN-Xyl)(NHC)2] (3) at ambient temperature and a dicarbodiimido complex trans-[Ni(NCN-Xyl)2(NHC)2] (4) at 70 °C. Reaction of alkyl isocyanides with 2 at ambient temperature gave the ditetrazolato complexes trans-[Ni(CN4-R)2(NHC)2] (5, R = tert-butyl; 6, R = cyclohexyl) in good yields. A novel cationic “abnormal” tetrazolin-5-ylidene complex trans-[Ni(CN4–tBu,Me)2(NHC)2](BF4)2 (7, tBu = tert-butyl) was synthesized by direct methylation of 5 with [Me3O]BF4. All compounds have been fully characterized by multinuclei NMR spectroscopies and ESI mass spectrometry. The solid state molecular structures of complexes 2, 4, 5, and 7•2CH2Cl2 have also been confirmed by X-ray diffraction studies.

35. H. V. Huynh*, R. Jothibasu “Formation of homoleptic tetracarbene versus cis chelating dicarbene complexes of Nickel(II) and applications in Kumada-Corriu couplings” Eur. J. Inorg. Chem. 2009, 1926–1931.
DOI: 10.1002/ejic.200801149
(invited contribution for a special issue on NHC chemistry)
The formation of mono- versus bis(chelate) Ni(II) complexes bearing N-heterocyclic dicarbene ligands can be controlled by the flexibility of the ligand bridge. A short methylene spacer exclusively gives rise to a dicationic bis(chelate) complex [Ni(MeCCmeth)2]Br2 (1), whereas a more flexible propylene spacer affords a neutral mono-chelate complex [NiBr2(MeCCprop)] (2). Complex 2 was found to autoionize very slowly to the corresponding dicationic bis(chelate) over ~45 days in d6-DMSO. The formation of bis versus mono-chelate can be attributed to the different stabilities of the resulting metallocycles. The catalytic activity of the mono-chelate 2 was tested in the Kumada-Corriu coupling of aryl halides with aryl-magnesium reagents at ambient temperature.

34. Y. Han, H. V. Huynh* “Mixed carbene-isocyanide Pd(II) complexes: synthesis, structures and reactivity towards nucleophiles” Dalton Trans. 2009, 2201-2209.
DOI: 10.1039/B816471A
Reaction of the bromo-bridged dimeric monocarbene complex [PdBr2(iPr2-bimy)]2 (1) with various isocyanides afforded a series of mixed carbene-isocyanide complexes [PdBr2(iPr2-bimy)(CN-R)] (2a: R = Cy; 2b: R = nBu; 2c: R = Xyl) as inseparable mixtures of trans– and cis-isomers. The reactivity of 2c towards nucleophiles was studied. A new mixed NHC-ADC Pd(II) complex (4) was obtained in low yield when 2c was reacted with 2,6-dimethylaniline. Reaction of 2c with hydrazine yielded a hydrazine-bridged complex (6) via ligand substitution. In addition, salt metathesis of 2c with AgO2C2F3 afforded a cis arranged complex [Pd(O2CCF3)2(iPr2-bimy)(CN-Xyl)] (cis–7). Most complexes were fully characterized by multinuclei NMR spectroscopies, ESI or FAB mass spectrometry and X-ray diffraction analysis.

33. S. K. Yen, L. L. Koh, H. V. Huynh, T. S. A. Hor* “Formation and structures of Pd(II) N,S-heterocyclic carbene-pyridyl mixed-ligand complexes” J. Organomet. Chem. 2009, 694, 332-338.
DOI: 10.1016/j.jorganchem.2008.10.048
Mononuclear mixed-ligand complexes of Pd(II) containing a N,S-heterocyclic carbene (NSHC) with a secondary alkyl N-substituent and pyridyl ligand, with the general formula [PdI2(C10H11NS)L] (C10H11NS = 3-isopropylbenzothiazolin-2-ylidene; L = pyridine, 2-aminopyridine, 3-iodopyridine and 4-tert-butyl-pyridine) have been synthesized and characterized by X-ray single-crystal crystallography. Both solution and solid-state structures, as evident from their 1H NMR spectra and X-ray structures, show anagostic γ-hydrogen interactions of metal with methine of the substituent on the carbene or pyridyl ligand giving 5-membered-chelate-like structures.

32. H. V. Huynh*, J. Wu “Rotamers of Palladium Complexes bearing IR active N-Heterocyclic Carbene ligands: Synthesis, Structural Characterization and Catalytic Activities” J. Organomet. Chem. 2009, 694, 323-331.
DOI: 10.1016/j.jorganchem.2008.10.044
(Ranked No. 5 of the top 25 hottest acrticles from January-March 2009 in Journal of Organometallic Chemistry)
The preparation and properties of mono- versus bis(carbene) Pd(II) complexes bearing unsymmetrical cyano- and ester-functionalized NHC-ligands as potential IR probes were studied in detail. Direct reaction of Pd(OAc)2 with functionalized imidazolium salts afforded either bis(carbene) (3a, c) or mono-carbene complexes (5, 6) with a N-coordinated imidazole co-ligand. The latter were exclusively obtained with N-ethylene substituted salts, which were found to undergo N–C cleavage reaction. The milder Ag-carbene transfer reaction on the other hand was tolerable to the length of the substituents and the nature of the functional groups. All bis(carbene) complexes (3a-c, 4a-c) were obtained as a inseparable mixture of square-planar trans-anti and trans-syn rotamers. The identity, ratio and dynamic equilibrium of these rotamers have been investigated and the relatively high rotational barrier for rotamers of 3a was estimated to be about 74 kJ mol-1 at 380 K. All eight complexes were fully characterized by NMR and IR spectroscopies, ESI mass spectrometry and X-ray single crystal and powder diffraction. A preliminary catalytic study showed that ester-functionalized complexes 4a and 4b gave rise to highly active catalyst in the double Mizoroki–Heck coupling of aryl dibromides, while the in situ ester-hydrolyzed complexes were also active in the coupling of activated aryl chlorides.

31. Y. Han, Y.-T. Hong, H. V. Huynh* “Ag(I) and Pd(II) Complexes of a 1,3-Dibenzhydryl Substituted Benzannulated N-Heterocyclic Carbene: Unexpected Rearrangement, Structures and Catalytic Studies” J. Organomet. Chem. 2008, 693, 3159-3165.
DOI: 10.1016/j.jorganchem.2008.06.037
Reaction of the sterically bulky 1,3-dibenzhydrylbenzimidazolium bromide (Bh2-bimyH+Br<sup-< sup=””>) (A) with Pd(OAc)2 in DMSO yielded a mono(carbene) Pd(II) complex (1) with a N-bound benzimidazole derivative, which resulted from an unusual NHC rearrangement reaction. Reaction of A with Ag2O, on the other hand, cleanly gave the Ag(I) carbene complex [AgBr(Bh2-bimy)] (2), which has been used as a carbene-transfer agent to prepare the acetonitrile complex trans-[PdBr2(CH3CN)(Bh2-bimy)] (3). Dissociation of acetonitrile from complex 3 and subsequent dimerization afforded the dinuclear Pd(II) complex [PdBr2(Bh2-bimy)]2 (4) in quantitative yield. All complexes were fully characterized by multinuclear NMR spectroscopies, ESI mass spectrometry and X-ray diffraction analysis. Furthermore, the catalytic activity of complex 4 in aqueous Suzuki-Miyaura cross-coupling reactions was studied and compared with that of its previously reported less bulky analogue [PdBr2(iPr2-bimy)]2.

30. S. K. Yen, L. L. Koh, H. V. Huynh, T. S. A. Hor* “Mono- and Dinuclear Palladium(II) N,S-Heterocyclic Carbene Complexes with N Spacers and their Suzuki Coupling Activities” Chem. Asian J. 2008, 3, 1649-1656.
DOI: 10.1002/asia.200800177
Mixed-ligand N,S-heterocyclic carbene (NSHC) complexes, trans-[PdBr2(NSHC)(Py)] (NSHC = 3-benzyl- or 3-propyl-benzothiazolin-2-ylidene), have been obtained from bridge-cleavage reactions of the dinuclear complex, [Pd(μ-Br)Br(NSHC)]2, in pyridine at room temperature. Use of neutral N-bidentate donors (L = pyrazine, 1,2-bis(4-pyridyl)ethane, 4,4’-bipyridine and trans-1,2-bis(4-pyridyl)-ethylene) yields the dinuclear spacer-bridged [Pd2Br4(NSHC)2(μ-L)] complexes. The X-ray single-crystal structures of the pyridyl, bridging pyrazine and 1,2-bis(4-pyridyl)ethane complexes are reported. These air-stable complexes are active in the Suzuki–Miyaura coupling reactions of selected aryl bromides. The dinuclear complexes are generally more active than their mononuclear pyridyl analogues. The benzyl derivatives consistently outperform the n-propyl counterparts.

29. A. T. Normand, S. K. Yen, H. V. Huynh, T. S. A. Hor, K. J. Cavell* “Catalytic Annulation of Heterocycles via a Novel Redox Process Involving the Imidazolium Salt N-Heterocyclic Carbene Couple” Organometallics 2008, 27, 3153-3160.
DOI: 10.1021/om800140n
A novel atom-efficient catalytic reaction, which converts imidazolium salts, with N-butenyl, N-substituted butenyl, and N-pentenyl substituents, into five- and six-membered fused-ring imidazolium and thiazolium salts has been developed. The reaction proceeds through azolium, C2-H, oxidative addition to Ni(0) followed by intramolecular insertion of the N-alkenyl double bond into the Ni hydride to give an intramolecularly bound carbene-Ni-alkyl intermediate. Reductive elimination of the linked carbene and alkyl groups gives the fused-ring azolium products and regenerates the Ni(0) catalyst. Products are potential building blocks for the synthesis of pharmaceuticals and novel ionic liquids. For example, 1,7-dimethyl-6,7-dihydro-5H-pyrrole[1,2-R]imidazolium bromide (2f), a five-membered fused-ring imidazolium salt, is formed from the catalytic ring closing of 1-butenyl-3-methylimidazolium bromide (1f). The reaction proceeds at moderate temperatures (50 °C) to give the products in high yield and selectivity. The catalyst was formed in situ from Ni(COD)2 plus added ligand L (where L = IMes, SMes, IPr, SPr, 4,5-Me2IPr, PPh3, PCy3, PCy2(Biphenyl), PtBu3) in DMF.

28. H. V. Huynh*, L. R. Wong, P. S. Ng “Anagostic Interactions and Catalytic Activities of Sterically Bulky Benzannulated N-Heterocyclic Carbene Complexes of Nickel(II)” Organometallics 2008, 27, 2231-2237.
DOI: 10.1021/om800004j
Nickel(II) bis(benzimidazolin-2-ylidene) complexes of the general formula [NiBr2(NHC)2] (NHC = 1,3-dibenzylbenzimidazolin-2-ylidene, 7; NHC = 1,3-diisopropylbenzimidazolin-2-ylidene, 8; NHC = 1,3-dibenzhydrylbenzimidazolin-2-ylidene, 9; NHC = 1,3-diisobutylbenzimidazolin-2-ylidene, 10; NHC = 1-isopropyl-3-benzylbenzimidazolin-2-ylidene, 11; NHC = 1-benzhydryl-3-benzylbenzimidazolin-2-ylidene, 12) have been prepared and fully characterized by spectroscopic methods and single-crystal X ray structure analyses. All complexes adopt a square-planar geometry with nickel as the crystallographic inversion center and a trans arrangement of the carbene ligands. For complexes 11 and 12 bearing unsymmetrically substituted ligands, only the trans-anti configuration was found in the solid state. In addition, the structures of 8, 9, 11 and 12 reveal a fixed orientation of the N-isopropyl and N benzhydryl substituents with the C-H groups pointing to the nickel(II) center to maximize rare intramolecular C-H⋅⋅⋅Ni anagostic or preagostic interactions. The large downfield shift of these C-H protons in the 1H NMR spectrum compared to their precursor salts indicates that these interactions are retained in solution. Preliminary catalytic studies show that complexes 7-12 are active in the Ullmann-coupling of bromobenzene and 4-bromoanisole. In particular, complexes 8, 9 and 12 with sterically more demanding ligands exhibit the best catalytic activities. The coupling reaction was found to be successful when carried out in neat [Bu4N]Br as ionic liquid, but not in dry DMF nor in DMF with [Bu4N]Br as an additive.

27. K. E. Neo, H. V. Huynh, L. L. Koh, W. Henderson, T.S. A. Hor* “Isolation and Crystallographic Characterization of Solvate- and Anion-Stabilized PCP Pincer Complexes of Palladium(II)” J. Organomet. Chem. 2008, 693, 1628-1635.
DOI: 10.1016/j.jorganchem.2007.11.031
Pincer PCP-Pd(II) complex [PdCl(PCP)] (1) (PCP = –CH(CH2CH2PPh2)2) reacts with AgNO3 to give [Pd(NO3)(PCP)] (2). Similar reaction with AgBF4 gives the aqua complex [Pd(OH2)(PCP)][BF4] (3) and the dinuclear complex [{Pd(PCP)}2(μ-Cl)][BF4] (4) with singly bridging chloro ligand. All new complexes were characterized by NMR spectroscopy, ESI-MS and single-crystal X-ray diffraction. Complex 1 and the triflate complex [Pd(OTf)(PCP)] (5) are active towards Suzuki–Miyaura coupling between aryl bromides and phenyl boronic acid.

26. S. K. Yen, L. L. Koh, H. V. Huynh*, T. S. A. Hor* “Pd(II) complexes with mixed benzothiazolin-2-ylidene and phosphine ligands and their catalytic activites toward C-C coupling reactions” Dalton Trans. 2008, 699-706.
DOI: 10.1039/b713152f
Novel Pd(II) mixed N,S-heterocyclic carbene (NSHC)-phosphine complexes of the general formula [PdBr2(NSHC)(PR3)] were obtained from bridge cleavage of dinuclear NSHC complexes of type [PdBr2(NSHC)]2 [NSHC = 3-benzylbenzothiazolin-2-ylidene and 3-propylbenzothiazolin-2-ylidene] with triphenylphosphine, tricyclohexylphosphine and 2-diphenylphosphanyl-pyridine. All complexes have been fully characterized by 1H and 13C NMR spectroscopy, ESI mass spectrometry and elemental analysis. The X-ray crystal structures of complexes 3–8 are reported. The complexes exhibit moderate to good catalytic activity in the Suzuki–Miyaura coupling reaction of aryl bromides and chlorides.

25. R. Jothibasu, H. V. Huynh*, L. L. Koh “Au(I) and Au(III) complexes of a sterically bulky benzimidazole-derived N-heterocyclic carbene” J. Organomet. Chem. 2008, 693, 374-380.
DOI: 10.1016/j.jorganchem.2007.11.003
(Ranked No. 3 of the top 25 hottest acrticles from January-March 2008 in Journal of Organometallic Chemistry)
The reaction of [AuCl(SMe2)] with in situ generated [AgCl(iPr2-bimy)] (iPr2-bimy = 1,3-diisopropylbenzimidazolin-2-ylidene), which in turn was obtained by the reaction of Ag2O with 1,3-diisopropylbenzimidazolium bromide (iPr2-bimyH+Br–, A), afforded the monocarbene Au(I) complex [AuCl(iPr2-bimy)] (1). Subsequent reaction of 1 and the ligand precursor iPr2-bimyH+BF4–, (B) in acetone in the presence of K2CO3 yielded the bis(carbene) complex [Au(iPr2-bimy)2]BF4 (2) as a white powder in 80% yield. The oxidative addition of elemental iodine to complex 2 gave the bis(carbene) Au(III) complex trans-[AuI2(iPr2-bimy)2]BF4 (3) as an orange-red powder in 92% yield. All complexes 1–3 have been fully characterized by multinuclear NMR spectroscopies, ESI mass spectrometry, elemental analysis, and X-ray single crystal diffraction. Complexes 1 and 2 adopt a linear geometry around metal centers as expected for d10 metals. The geometry around the Au(III) metal center in 3 is essentially square-planar with two carbene ligands in trans-position to each other. Complex 3 shows absorption and photoluminescence properties owing to a ligand to metal charge transfer.
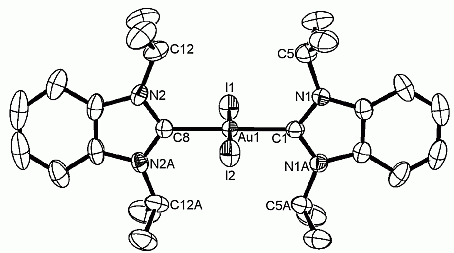
24. H. V. Huynh*, R. Jothibasu, L. L. Koh “Dipalladium bis(µ-isopropylthiolato) complexes with a [Pd2S2] core supported by N-heterocyclic carbenes” Organometallics 2007, 26, 6852-6856.
DOI: 10.1021/om700867e
The reaction of Pd(OAc)2 with 1,3-dibenzylbenzimidazolium bromide (A) and 1-propyl-3-methylbenzimidazolium iodide (B) afforded the dihalo-bis(carbene) complexes cis-[PdBr2(Bz2-bimy)2] (1) and cis-[PdI2(Pr,Me-bimy)2] (2), respectively. Halide substitution of 1 and 2 with AgO2CCH3 gave the mixed diacetato-bis(carbene) complexes cis-[Pd(O2CCH3)2(Bz2-bimy)2] (3) and cis-[Pd(O2CCH3)2(Pr,Me-bimy)2] (4). In situ deprotonation of isopropylthiol with the mixed carbene-carboxylato complexes 3 and 4 yielded the novel dipalladium complexes [Pd2(μ–iPr-S)2(Bz2-bimy)4](BF4)2 (5) and [Pd2(μ–iPr-S)2(Pr,Me-bimy)4](BF4)2 (6) with a [Pd2S2] core solely supported by N-heterocyclic carbenes. All compounds have been fully characterized by multinuclei NMR spectroscopies and ESI mass spectrometry. The solid state molecular structures of complexes 2, 3, 5 and 6 have also been confirmed by X-ray diffraction studies.

23. Y. Han, H. V. Huynh*, G. K. Tan “Palladium(II) Pyrazolin-4-ylidenes: Remote N-Heterocyclic Carbene Complexes and Their Catalytic Application in Aqueous Suzuki-Miyaura Coupling” Organometallics 2007, 26, 6581-6585.
DOI: 10.1021/om7009107
(Ranked No. 6 of the most-accessed articles in Oct-Dec 2007 in Organometallics)
Two mono-cationic complexes of pyrazole-derived remote carbene ligands of the type trans [PdI(rNHC)(PPh3)2]+OTf– {rNHC = 2-ethyl-3,5-dimethyl-1-phenylpyrazolin-4-ylidene (6a); rNHC = 1-ethyl-2,3,5-trimethylpyrazolin-4-ylidene (6b)}, were prepared by oxidative addition of 4 iodo-1,2,3,5-tetrasubstituted pyrazolium triflate salts to [Pd2(dba)3]/PPh3 in good yields. Both compounds were fully characterized by multinuclei NMR spectroscopies, ESI mass spectrometry and X-ray diffraction analysis. A comparative study on the aqueous Suzuki-Miyaura catalytic activities of these cationic complexes with their previously reported neutral counterparts reveals the superiority of the former.
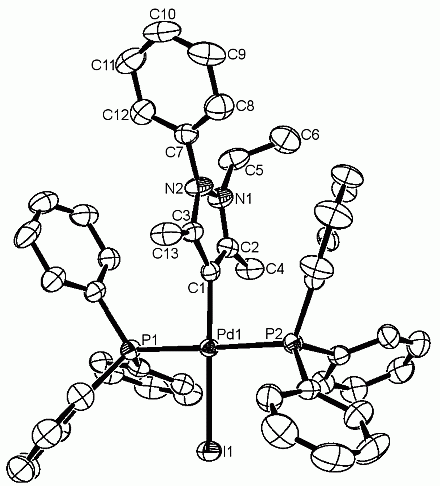
22. Y. Han, H. V. Huynh*, G. K. Tan “Syntheses and Characterizations of Pd(II) Complexes incorporating a N-Heterocyclic Carbene and Aromatic N-Heterocycles” Organometallics 2007, 26, 6447-6452.
DOI: 10.1021/om700753d
Reaction of the bromo-bridged dimeric monocarbene complex [PdBr2(iPr2-bimy)]2 (1) with various bidentate N-heterocycles afforded novel linear dinuclear Pd(II) complexes {[PdBr2(iPr2-bimy)]2(μ-L)} {μ-L = 4,4’-bipyridine (2); μ-L = 4,4’-bipyridylethane (3); μ-L = 4,4’-bipyridylethylene (4)}. The mononuclear counterpart [PdBr2(iPr2-bimy)(Pyridine)] (5) has also been synthesized by reaction of 1 with pyridine. All compounds have been fully characterized by multi nuclei NMR spectroscopies, FAB mass spectrometry and X-ray diffraction analyses. Molecular structures of 2-5 show a fixed orientation of the C-H protons in the N-isopropyl substituents towards the metal center suggesting interesting C-H⋅⋅⋅Pd preagostic interactions. These interactions are retained in solution as indicated by the large downfield shift of these C-H protons in the 1H NMR spectrum. UV-Vis and CV studies revealed that the dinuclear complexes 2-4 have similar electrochemical behavior to the mononuclear species 5.

21. K. E. Neo, H. V. Huynh, L. L. Koh, W. Henderson, T. S. A. Hor* “Dinuclear PCP pincer complexes from Lewis acidic [Pd(OTf)(PCP)] and basic [Pd(4-Spy)(PCP)] (OTf = triflate; 4-Spy = 4-pyridinethiolate; PCP = –CH(CH2CH2PPh2)2)” Dalton Trans. 2007, 5701-5709.
DOI: 10.1039/b710752h
The Lewis acidic pincer with a labile triflate ligand, viz. [Pd(OTf)(PCP)] (PCP = –CH(CH2CH2PPh2)2) 1 was prepared from [PdCl(PCP)] with AgOTf. It reacts readily with neutral bidentate ligands [L = 4,4′-bipyridine (4,4′-bpy) and 1,1-bis(diphenylphosphino)ferrocene (dppf)] to give dinuclear PCP pincers [{Pd(PCP)}2(μ-L)][OTf]2 (L = 4,4′-bpy, 2; dppf, 3). [PdCl(PCP)] also reacts with 4-mercaptopyridine in the presence of KOH to give a Lewis basic pincer with a free pyridine functional group [Pd(4-Spy)(PCP)] 4. Its metalloligand character is exemplified by the isolation of an asymmetric dinuclear double-pincer complex [{Pd(PCP)}2(μ-4-Spy)][PF6] 6 bridged by an ambidentate pyridinethiolato ligand. Complexes 1, 2, 3, 4 and 6 have been characterized by single-crystal X-ray diffraction analyses.

20. Y. Han, H. V. Huynh*, G. K. Tan “Mono- vs Bis(carbene) Complexes: A Detailed Study on Platinum(II)-Benzimidazolin-2-ylidenes” Organometallics 2007, 26, 4612-4617.
DOI: 10.1021/om700543p
The reaction of PtBr2 with NaOAc and 1,3-diisopropylbenzimidazolium bromide (A) in DMSO afforded the mixed monocarbene-DMSO complex cis-[PtBr2(DMSO)(iPr2-bimy)] (cis–1) and the bis(carbene) complex trans-[PtBr2(iPr2-bimy)2] (trans–2). The DMSO ligand in cis–1 can be easily replaced by stronger donors such as triphenylphosphine and pyridine to give novel benzannulated monocarbene complexes trans-[PtBr2(iPr2-bimy)(PPh3)] (trans–3), cis-[PtBr2(iPr2-bimy)(PPh3)] (cis–3) and trans-[PtBr2(iPr2-bimy)(Pyridine)] (trans–4), respectively. All compounds have been fully characterized by multinuclei NMR spectroscopies and mass spectrometry (ESI, FAB). X-ray diffraction studies on single crystals of cis–1, trans–2, cis–3 and trans–4 revealed a square planar geometry and a fixed orientation of the N-isopropyl substituents with the C-H protons pointing to the metal center to maximize interesting and rare C-H⋅⋅⋅Pt preagostic interactions. These interactions are also retained in solution as indicated by the large downfield shift of the isopropyl C-H protons in the 1H NMR spectrum compared to that in the precursor salt A.

19. S. K. Yen, L. L. Koh, H. V. Huynh*, T. S. A. Hor* “Pd(II) complexes of N,S-heterocyclic carbenes with pendant and coordinated allyl function and their Suzuki coupling activities” Dalton Trans. 2007, 3952-3956.
DOI: 10.1039/B706968E
3-(2-propenyl)benzothiazolium bromide (A) provided a direct and simple entry to Pd(II) complexes with N,S-heterocyclic carbene (NSHC) ligands functionalized with an allyl pendant with hemilabile potential. Addition of salt A to Pd(OAc)2 eliminated HOAc and afforded the bis(carbene) complexes cis [PdBr2(NHSC)2] (cis–1) (NSHC = 3-(2-propenyl)benzothiazolin-2-ylidene) and trans [PdBr2(NHSC) 2] (trans–1) along with the monocarbene complexes [PdBr2(NSHC)] (2) and trans-[PdBr2(NSHC)(benzothiazole-κN)] (3) as minor side products. Salt-metathesis of cis–1 with AgO2CCF3 yielded the mixed dicarboxylato-bis(carbene) complex cis-[Pd(O2CCF3)2(NSHC)2](4). Complexes cis–1, trans–1 and 4 were characterized by multinuclear NMR spectroscopies, ESI mass spectrometry and elemental analysis. The molecular structures of complexes cis–1, 2 and 3 have been determined by X-ray single crystal diffraction. Complexes cis–1 and 4 as well as a in situ mixture of Pd(OAc)2 and salt A were found to be active towards Suzuki-Miyaura coupling of aryl bromides and activated aryl chlorides giving good conversions.

18. Y. Han, H. V. Huynh*, L. L. Koh “Pd(II) Complexes of a Sterically Bulky, Benzannulated N-Heterocyclic Carbene and Their Catalytic Activities in the Mizoroki-Heck Reaction” J. Organomet. Chem. 2007, 692, 3606-3613.
DOI: 10.1016/jorganchem.2007.04.037
(Ranked No. 2 of the top 25 hottest acrticles from July-September 2007 inJournal of Organometallic Chemistry)
The bis(N,N’-diisopropylbenzimidazolin-2-ylidene)Pd(II) complexes trans-[PdBr2(iPr2-bimy)2] (trans–1) and trans-[PdI2(iPr2-bimy)2] (trans–2) have been prepared in good yields by in situ deprotonation of the corresponding N,N’-diisopropylbenzimidazolium salt (iPr2-bimyH+X-) (A: X = Br, B: X = I) with Pd(OAc)2 in DMSO at elevated temperature. Salt metathesis of trans–1 or trans–2 with AgO2CCF3 in refluxing CH3CN afforded the novel mixed carbene-carboxylato complex cis-[Pd(O2CCF3)2(iPr2-bimy)2] (cis–3). This halo/trifluorocarboxylato ligand substitution can be regarded as a selective method for the synthesis of cis-configured bis(carbene) complexes. All compounds have been fully characterized by multinuclei NMR spectroscopies and ESI mass spectrometry. X-ray diffraction studies on single crystals of trans–1, trans–2 and cis–3 revealed a square planar geometry and a fixed orientation of the N-isopropyl substituents with the C-H protons pointing to the metal center to maximize rare C-H⋅⋅⋅Pd preagostic interactions. These interactions are also retained in solution as indicated by the large downfield shift of the isopropyl C-H protons in the 1H NMR spectrum compared to those in precursor salts A or B. A preliminary catalytic study revealed that all complexes are highly active in the Mizoroki-Heck coupling of aryl bromides and chlorides. However, these complexes gave slower conversions as compared to catalysts with less bulky benzimidazolin-2-ylidenes. This is most likely due to the steric bulk of the ligands, which hamper a fast reductive formation of catalytically active Pd(0) species.

17. K. E. Neo, Y. Y. Ong, H. V. Huynh, T. S. A. Hor “A single-molecular pathway from heterometallic MM (M = BaII, MnII; M = CrIII) oxalato complexes to intermetallic composite oxides” J. Mater. Chem. 2007, 17, 1002-1006.
DOI: 10.1039/b609630a
Thermolysis of {(n-C4H9)4N[MnIICrIII(C2O4)3]}n, 1 at 500 °C for 10 h gives spinel Mn1.5Cr1.5O4 which was characterized by powder XRD, SEM, FTIR and elemental analysis. The thermal conversion occurs by an internal redox process at ca. 400 °C in one step (TGA). It offers an alternative molecular source for an intermetallic oxide. Thermolysis of {[BaII6 (H2O)17][CrIII(C2O4)3]4}⋅ 7H2O, 2 under similar conditions gives a mixture of BaIICrVIO4 and BaIICO3. These results suggested that the oxalate ligand in a heterometallic complex may be a convenient source for oxides in intermetallic composites and that, under suitable conditions, both metals in the heterometallic complexes can be transferred to the intermetallic oxides. It suggested that composite metal oxides can be generated from hetero- and inter-metallic oxalato complexes at relatively low temperatures, which could serve as a convenient route for the preparation of technologically important composites.

16. Y. Han, H. V. Huynh* “Preparation and characterization of the first pyrazole-based remote N-heterocyclic carbene complexes of palladium(II)” Chem. Commun. 2007, 1089-1091.
DOI: 10.1039/b615441g
The first pyrazolin-4-ylidene complexes of palladium(II) have been synthesized by oxidative addition of 4-iodopyrazolium salts to Pd2(dba)3/PPh3 and were fully characterized by multinuclear NMR spectroscopies, ESI mass spectrometry and X-ray diffraction studies.

15. T. Kreikmann, C. Diedrich, T. Pape, H. V. Huynh, S. Grimme, F. E. Hahn* “Metallosupramolecular Chemistry with Bis(benzene-o-dithiolato) Ligands” J. Am. Chem. Soc. 2006, 128, 11808-11819.
DOI: 10.1021/ja063655u
The bis(benzene-o-dithiol) ligands H4–1, H4–2 and H4–3 react with [Ti(OC2H5)4] to give dinuclear triple-stranded helicates [Ti2L3]4- (L = 14-, 24-, 34-). NMR spectroscopic investigations revealed that the complex anions possess C3 symmetry in solution. A crystal structure analysis for (PNP)4[Ti2(2)3] ((PNP)4[14]) confirmed the C3 symmetry for the complex anion in the solid state. The complex anion in Li(PNP)3[Ti2(1)3] (Li(PNP) 3[13]) does not exhibit C3 symmetry in the solid state due to the formation of polymeric chains of lithium bridged complex anions. Complexes [13]4- and [14]4- were obtained as racemic mixtures of the Δ,Δ and Λ,Λ isomers. In contrast to that, complex (PNP)4[Ti2(3)3] ((PNP)4[15]) with the enantiomerically pure chiral ligand 34- shows a strong Cotton effect in the CD spectrum, indicating that the chirality of the ligands leads to the formation of chiral metal centers. The o-phenylene diamine bridged bis(benzene-o-dithiol) ligand H4–4 reacts with Ti4+ to give the dinuclear double-stranded complex Li2[Ti2(4)2(μ-OCH3)2] containing two bridging methoxy ligands between the metal centers. The crystal structure analysis and the 1H NMR spectrum of (Ph4As)2[Ti2(4)2(μ-OCH3)2] ((Ph4As)2[(16]) reveal C2 symmetry for the anion [Ti2(4) 2(μ-OCH3)2]2-. For a comparative study the dicatechol ligand H4–5, containing the same o-phenylene diamine bridging group as the bis(benzene-o-dithiol) ligands H4–4 was prepared and reacted with [TiO(acac)2] to give the dinuclear complex anion [Ti2(5) 2(μ-OCH3)2]2-. The molecular structure of (PNP)2[Ti2(5)2(μ-OCH3)2] ((PNP)2[17]) contains a complex anion which is similar to [16]2-, with the exception that strong N-H⋅⋅⋅O hydrogen bonds are formed in complex anion [17]2-, while N-H⋅⋅⋅S hydrogen bonds are absent in complex anion [16]2-.

14. S. K. Yen, L. L. Koh, F. E. Hahn, H. V. Huynh*, T. S. A. Hor* “Convenient Entry to Mono- and Dinuclear Palladium(II) Benzothiazolin-2-ylidene Complexes and their Activities towards Heck-Coupling” Organometallics 2006, 25, 5105-5112.
DOI: 10.1021/om060510n
Under solventless conditions, benzothiazole reacts with benzyl bromide to give near-quantitative yield of the salt N-benzylbenzothiazolium bromide (A), which is a convenient air-stable heterocyclic carbene precursor. Treatment of A with Pd(OAc)2 in CH3CN affords the bis(carbene) complex cis-[PdBr2(NHC)2] (1) (NHC = N-benzylbenzothiazolin-2-ylidene). In DMSO, this reaction yields an unprecedented dinuclear N,S-heterocyclic carbene complex, [PdBr2(NHC)]2 (2). Complex 2 undergoes bridge cleavage reactions with CH3CN and DMF to give the mononuclear and solvated monocarbene complexes trans-[PdBr2(NHC)(Solv)] [Solv = CH3CN (3) and DMF (4)]. All compounds have been fully characterized by 1H and 13C NMR spectroscopy, ESI or FAB mass spectrometry, and elemental analysis. The molecular structures of A and 1-4 have been determined by X-ray single-crystal diffraction. The catalytic activities of 1-4 toward Mizoroki-Heck coupling reactions of aryl bromides with tert-butyl acrylate are described and compared.

13. H. V. Huynh*, C. H. Yeo, G. K. Tan “Hemilabile Behavior of a Thioether-functionalized N-Heterocyclic Carbene ligand” Chem. Commun. 2006, 3833-3835.
DOI: 10.1039/b608325k
The truely hemilabile nature of a novel thioether-functionalized N-heterocyclic carbene ligand is demonstrated in a range of Pd(II) complexes.

12. H. V. Huynh*, Y. Han, J. H. H. Ho, G. K. Tan “Palladium(II) Complexes of a sterically bulky, benzannulated N-Heterocyclic Carbene with unusual C-H⋅⋅⋅Pd and Ccarbene⋅⋅⋅Br interactions” Organometallics 2006, 25, 3267-3274.
DOI: 10.1021/om060151w
(Ranked No. 9 of the most-accessed articles in Apr-Jun 2006 in Organometallics)
The sterically bulky carbene precursor 1,3-diisopropylbenzimidazolium bromide (iPr2-bimyH+Br-) (A) has been prepared by an improved method in 84% yield. Reaction of A with Pd(OAc)2 and NaBr gave the dimeric Pd(II) benzimidazolin-2-ylidene complex [PdBr2(iPr2-bimy)]2 (1), which can be easily cleaved by CH3CN, another equivalent of salt A, and triphenylphosphine to afford the novel benzannulated monocarbene complexes trans-[PdBr2(CH3CN)( iPr2-bimy)] (2), (iPr2-bimyH)[PdBr3(iPr2-bimy)] (3), trans-[PdBr2(iPr2-bimy)(Ph3P)] (trans–4), and cis-[PdBr2(iPr2-bimy)(Ph3P)] (cis–4), respectively. All compounds have been fully characterized by multinuclei NMR spectroscopies and mass spectrometries (FAB, ESI). X-ray diffraction studies on single crystals of 1-3 and cis–4 revealed a square planar geometry and a fixed orientation of the N-isopropyl substituents with the C-H group pointing to the metal center to maximize C-H⋅⋅⋅Pd interactions. The large downfield shift of the C-H protons in the 1H NMR spectrum compared to the precursor A indicates that these C-H⋅⋅⋅Pd interactions are retained in solution and better described as weak hydrogen bonds, rather than as agostic interactions. Furthermore, the molecular structures of especially complexes 2 and 3 clearly show a bending of the bromo ligands toward the carbene carbon atom in order to maximize intramolecular Ccarbene⋅⋅⋅Br interactions. The nature of these interactions can be attributed to a form of back-bonding to the formally vacant p-orbital of the Ccarbene atom with the electron density originating from the bromo ligands’ lone pairs. A detailed study on the trans-cis isomerization of the mixed NHC-phosphine complexes 4 revealed that a cis arrangement in such complexes is thermodynamically favored. Furthermore, a preliminary catalytic study shows that complex 1 is highly active in the Suzuki-Miyaura coupling of aryl bromides and chlorides in pure water as environmentally benign solvent.

11. H. V. Huynh*, N. Meier, T. Pape, F. E. Hahn* “Benzothiazolin-2-ylidene Complexes of Iridium(I)“ Organometallics 2006, 25, 3012-3018.
DOI: 10.1021/om060006i
The reaction of allyl bromide with benzothiazole under neat conditions furnished 3-(2-propenyl)-benzothiazolium bromide, (H-1)Br, in high yield. Attempts to synthesize the corresponding carbene dimer by deprotonation of (H-1)+ led to the isolation of the rearrangement product 2,3-di(2-propenyl)-2’,3’-dihydro-2,2’-bisbenzothiazole (2). The reaction of [Ir(μ-OMe)(cod)]2 with the salt (H-1)Br unexpectedly afforded [IrBr(cod)(benzothiazole)] (3) (cod = 1,5-cyclooctadiene), which contained an N-coordinated unsubstituted benzothiazole ligand. The formation of carbene complexes of IrI with the benzothiazolin-2-ylidene ligand could be achieved via precoordination of the allyl substituent of (H-1)+ to [Ir(cod)(MeCN)2]BF4 and subsequent deprotonation at the C2 position of (H-1)+ by addition of base. The use of NaH as external base yielded the square planar IrI complex 4 with an N-propyl-substituted carbene ligand, while deprotonation with KOtBu gave the five-coordinated IrI complex [IrBr(cod)(η2–1)], 5. Displacement of the cod ligand in complex 4 by two CO ligands afforded the complex [IrBr(CO)2(NHC)] (NHC = 3-propylbenzothiazolin-2-ylidene), 6, which allowed an estimation of the σ-donor capabilities of benzothiazolin-2-ylidene ligands. Compounds 1-6 have been characterized spectroscopically, and the molecular structures of (H-1)Br and 2-5 were determined by X-ray diffraction.

10. H. V. Huynh*, T. C. Neo, G. K. Tan “Mixed Dicarboxylato-Bis(carbene) Complexes of Palladium(II): Synthesis, Structures, trans-cis Isomerism and Catalytic Activity” Organometallics 2006, 25, 1298-1302.
DOI: 10.1021/om0510369
(Ranked No. 13 of the most-accessed articles in Jan-Mar 2006 in Organometallics; Cited in Annual Reports for 2006: R. Singh, S. P. Nolan “N-Heterocyclic carbenes: Advances in transition metal and organic catalysis” Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2006, 102, 168.)
Mixed dicarboxylato-bis(carbene) complexes of palladium(II) have been prepared by reacting cis-diiodo-bis(N,N’-dimethylbenzimidazolin-2-ylidene)palladium(II) (cis–A) with AgO2CR, where R ) CH3, CF3, and CF2CF3. In this manner, cis-diacetato-bis(N,N’-dimethylbenzimidazolin-2-ylidene)palladium(II) (1), cis-di(trifluoroacetato)-bis(N,N’-dimethylbenzimidazolin-2-ylidene)palladium(II) (2), and cis-di(pentafluoro-propionato)-bis(N,N’-dimethylbenzimidazolin-2-ylidene)palladium(II) (3) were obtained. Complexes 1-3 were fully characterized by multinuclear NMR spectroscopies as well as ESI mass spectrometry. X-ray crystal structure analyses of the first mixed carboxylato/benzimidazolin-2-ylidene complexes reveal mononuclear species with a square-planar palladium(II) center coordinated by two monodentate carbene and two monodentate carboxylato ligands in a cis arrangement. The cis configuration was found to be thermodynamically favored in this type of complexes. The results also show that the introduction of N-heterocyclic carbene ligands stabilizes the palladium-carboxylate moiety effectively, thus preventing both reductive decomposition and autoionization processes. A preliminary catalytic study revealed that complexes 1-3 are highly active in the Mizoroki-Heck coupling of aryl bromides and activated aryl chlorides.

9. H. V. Huynh*, C. Holtgrewe, T. Pape, L. L. Koh, E. Hahn* “Synthesis and Structural Characterization of the First Bis(benzimidazolin-2-ylidene) Complexes of Nickel(II)” Organometallics 2006, 25, 245-249.
DOI: 10.1021/om050781i
The reaction of Ni(OAc)2 with the benzimidazolium salts 1,3-dimethylbenzimidazolium iodide (1), 1,3-bis(2-propenyl)benzimidazolium bromide (2), 1,3-dipropylbenzimidazolium bromide (3), 1-(2-propenyl)-3-methylbenzimidazolium bromide (6), and 1-propyl-3-methylbenzimidazolium iodide (7) in molten [Bu4N]X (X = Br, I, BF4) as ionic liquid afforded novel square-planar nickel(II) bis(benzimidazolin-2-ylidene) complexes of the general formula trans-[NiX2(NHC)2] (X = I, NHC = 1,3-dimethylbenzimidazolin-2-ylidene, 8; X = Br, NHC = 1,3-bis(2-propenyl)benzimidazolin-2-ylidene, 9; X = Br, NHC = 1,3-dipropylbenzimidazolin-2-ylidene, 10; X = Br, NHC = 1-(2-propenyl)-3-methylbenzimidazolin-2-ylidene, 11; X = I, NHC = 1-propyl-3-methylbenzimidazolin-2-ylidene, 12). X-ray diffraction studies of 8-12 reveal a square-planar coordination geometry for all complexes with the carbene ligands arranged in a trans fashion.

8. S. K. Quek, I. Lyapkalo*, H. V. Huynh “A New Highly Efficient Method for the Synthesis of tert-Alkyl Nitroso Compounds” Synthesis 2006, 1423-1426.
DOI: 10.1055/s-2006-926442
The syntheses of tert-alkyl nitroso compounds RCH2CMe2N=O from commercially available tert-alkyl amines RCH2CMe2NH2 proceed cleanly via the intermediacy of the benzoyl derivatives RCH2CMe2NHOC(O)Ph and the corresponding hydroxylamines RCH2CMe2NHOH. Since the intermediates require no purification in the course of the transformations, the overall yields of the isolated crystalline nitroso dimers (75–80% for R = H, 75% for R = Me and 66% for R = Me3C) are based on the corresponding amine precursors. In the latter case (R = Me3C), significant steric demands and hydrophobicity of Me3CCH2CMe2 group necessitate the application of more efficient reagents and conditions on the debenzoylation and oxidation steps. The syntheses are perfectly suitable for scale-up and were successfully performed on up to 500-mmol scale.
7. S. K. Quek, I. Lyapkalo*, H. V. Huynh “Synthesis and Properties of N,N’-dialkyl-imidazolium bis(nonafluorobutane-1-sulfonyl)imides: a new subfamily of ionic liquids” Tetrahedron 2006, 62, 3137-3145.
DOI: 10.1016/j.tet.2006.01.015
A series of N,N’-dialkylimidazolium bis(nonafluorobutane-1-sulfonyl)imides was synthesized in high yields by quaternization of imidazole derivatives with various readily available alkylating reagents, followed by anion exchange with highly stable and non-hygroscopic potassium bis(nonafluorobutane-1-sulfonyl)imide. The latter was obtained by an improved method starting from ammonium chloride and nonafluorobutane-1-sulfonyl fluoride. The quaternary imidazolium salts thus obtained constitute a new subfamily of thermally stable and remarkably hydrophobic ionic liquids with melting points in the range 0–40 °C and solubilities in water and organic solvents (aromatic hydrocarbons, dialkyl ethers) in the range of 0.5–1.5 wt%. The ionic liquids can be easily purified from ionic byproducts (e.g., halogenide salts) by aqueous extraction followed by thorough drying in a high vacuum without loss of yield. Due to the above features, these new ionic fluids may be considered as promising recyclable media in repeated catalytic processes.
6. H. V. Huynh*, J. H. H. Ho, T. C. Neo, L. L. Koh “Solvent-controlled selective Synthesis of a trans-configured Benzimidazoline-2-ylidene Palladium(II) Complex and Investigations of its Heck-type catalytic activity” J. Organomet. Chem. 2005, 690, 3854-3860.
DOI: 10.1016/j.jorganchem.2005.04.053
(Among the top 50 most cited articles from 2005-2008 in Journal of Organometallic Chemistry)
Reaction of N,N’-dimethylbenzimidazolyl iodide A with Pd(OAc)2 in DMSO gives selectively trans-bis(N,N’-dimethylbenzimidazoline-2-ylidene) palladium(II) diiodide (trans–2) in 77% yield. The selective formation of the trans-coordination isomer and thus the cis-trans rearrangement is driven by the insolubility of trans–2 in DMSO. X-ray single-crystal diffraction analysis and 13C NMR spectroscopy confirm the trans-geometry of the square planar Pd(II) complex. Catalytic studies show that cis–1 and trans–2 are highly efficient in the Mizoroki-Heck coupling reaction of aryl bromides and activated aryl chlorides both in DMF and [N(n-C4H9)4]Br as ionic liquid. The catalytic activities of Pd(II) complexes with N-heterocyclic carbene ligands derived from benzimidazole are comparable to their imidazole-derived analogues.

5. H. V. Huynh*, D. LeVan, F. E. Hahn*, T. S. A. Hor “Synthesis and Structural Characterization of mixed Carbene-Carboxylate Complexes of Palladium(II)” J. Organomet. Chem. 2004, 689, 1766-1770.
DOI: 10.10.16/j.jorganchem.2004.02.033
Mixed carbene-carboxylate complexes of Palladium(II) have been prepared by reacting {1,1’-dimethyl-3,3’methylene-diimidazoline-2,2’-diylidene}palladium(II) diiodide 1 with AgO2CR, where R = CF3, CF2CF3 and CF2CF2CF3. In this manner, {1,1’-dimethyl-3,3’methylenediimidazoline-2,2’-diylidene}palladium(II) bis(trifluoroacetate) (2), {1,1’-dimethyl-3,3’methylenediimidazoline-2,2’-diylidene}palladium(II) bis(pentafluoropropionate) (3) and {1,1’-dimethyl-3,3’methylenedi-imidazoline-2,2’-diylidene}palladium(II) bis(heptafluorobutyrate) (4) were obtained. All three complexes were fully characterized by 1H-, 13C- and 19F-NMR spectroscopy as well as ESI mass spectrometry. X-ray crystal structure analyses of complexes 3 and 4 reveal mononuclear species with a square planar metal center coordinated by a cis-chelating dicarbene and two monodentate carboxylate ligands. The results show that the introduction of a cis-chelating N,N-heterocyclic carbene ligand stabilizes the palladium-carboxylate moiety effectively.
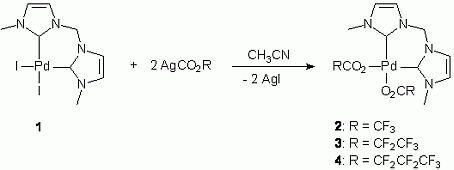
4. H. V. Huynh, R. Fröhlich, F. E. Hahn* “Synthesis and Molecular Structure of [Fe2Cl2(µ-S-t-Bu)2(η1–η1–µ-dppe)]” Z. Naturforsch. 2003, 58b, 359-361.
Free full text available: http://www.znaturforsch.com/ab/v58b/s58b0359.pdf
The red title compound was synthesized by the metathesis reaction of [FeCl2(dppe)] (dppe = Ph2P(CH2)2PPh2) and NaS-tBu in THF. The X-ray structure analysis revealed a dinuclear complex with two iron(II) centers coordinated in a distorted tetrahedral fashion by two bridging thiolates, one bridging dppe molecule and one terminal chloro ligand.
3. H. V. Huynh, W. W. Seidel, T. Lügger, R. Fröhlich, B. Wibbeling, F. E. Hahn* “ortho-Lithiation of Benzene-1,2-dithiol: A Methodology for ortho-Functionalization of Benzene-1,2-dithiol” Z. Naturforsch. 2002, 57b, 1401-1408.
Free full text available: http://www.znaturforsch.com/ab/v57b/s57b1401.pdf
The ortho-lithiation of benzene-1,2-dithiol with three equivalents of nBuLi afforded mono- as well as di-C-lithiated intermediates. The surprising kinetically controlled formation of di-C-lithiated species offers the opportunity to obtain dimercaptobenzoic and -terephthalic acid derivatives in a one pot synthesis in reasonable yields. Both compounds are versatile building blocks for the synthesis of macrocycles containing arylenedithiol units. Focusing on novel terephthalic acid derivatives, two preparation routes, via amide and via alkyl linkage, respectively, have been elaborated. The reaction sequence (i) sulfur protection using either isopropyl or benzyl groups, (ii) transformation of the carboxylic function into an amide or an alkyl group and (iii) subsequent removal of the protection groups afforded the terephthaldiamidedithiol 6 and the 1,2-bis(2,3-dimercaptophenyl)ethane 11.
2. H. V. Huynh, T. Lügger, F. E. Hahn* “Synthesis and X-ray Molecular Structure of [WVI(C6H4S2-1,2)3] Completing the Structural Characterization of the Series [W(C6H4S2-1,2)3]n- (n = 0, 1, 2): Trigonal-Prismatic versus Octahedral Coordination in Tris(benzene-1,2-dithiolate) Complexes” Eur. J. Inorg. Chem. 2002, 3007-3009.
DOI: 10.1002/1099-0682(200211)2002:11<3007::AID-EJIC3007>3.0.CO;2-I
The tris(benzene-1,2-dithiolato) complex [WVI(C6H4S2-1,2)3] 1 was synthesized from [W(CH3)6] and C6H4(SH)2-1,2 in diethyl ether. Crystals of [WVI(C6H4S2-1,2)3] were obtained from a saturated dichloromethane solution at room temperature. The X-ray crystal structure analysis revealed that the tungsten atom in 1 is coordinated in an almost perfect trigonal-prismatic fashion with W-S distances between 2.3724(14) Å and 2.3840(14) Å.
1. H. V. Huynh, C. Schulze-Isfort, W. W. Seidel, T. Lügger, R. Fröhlich, O. Kataeva, F. E. Hahn* “Dinuclear Complexes with Bis(benzenedithiolate) Ligands” Chem. Eur. J. 2002, 8, 1327-1335.
DOI: 10.1002/1521-3765(20020315)8:6<1327::AID-CHEM1327>3.0.CO;2-N
As a part of a broader study directed towards helical coordination compounds with benzenedithiolate donors, we have synthesized the bis(benzenedithiol) ligands 1,2-bis(2,3-dimercaptobenzamido)ethane (H4–1) and 1,2-bis(2,3-dimercaptophenyl)ethane (H4–2). Both ligands form dinuclear complexes with NiII, NiIII and, after air-oxidation, CoIII ions under equilibrium conditions. Complexes (NEt4)4[NiII2(1)2] (11b), (NEt4)2[NiIII2(1)2] (13), and Na4-[NiII2(2)2] (14) were characterized by X-ray diffraction. In all complexes, two square-planar [Ni(S2C6H3R)2] units are linked in a double-stranded fashion by the carbon backbone and they assume a coplanar arrangement in a stairlike manner. Cyclic voltammetric investigations show a strong dependence of the redox potential on the type of the ligand. The substitution of 14- for 24- on nickel (-785 mV for 11b versus -1130 mV for 14, relative to ferrocene) affects the redox potential to a similar degree as the substitution of nickel for cobalt (-1160 mV for [Co2(1)2]2-/[Co2(1)2]4-, relative to ferrocene). The redox waves display a markedly less reversible behavior for complexes with the shorter bridged ligand 24- compared to those of 14-.

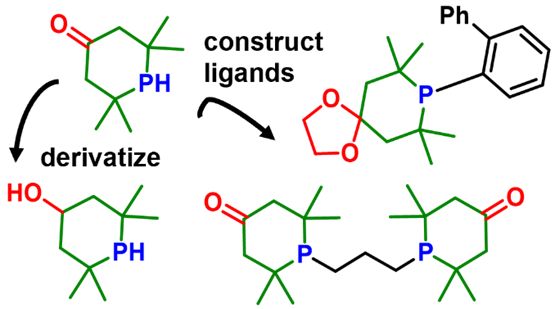
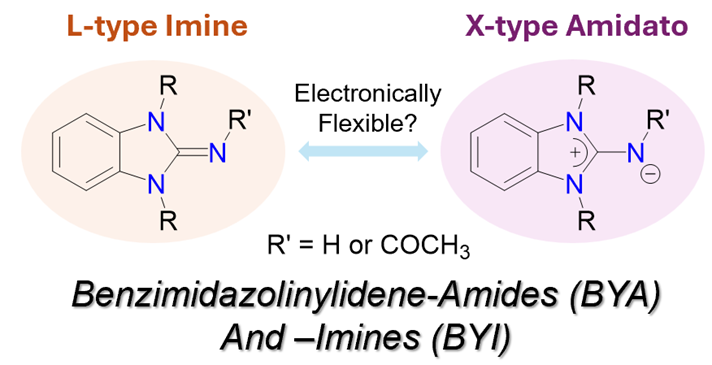
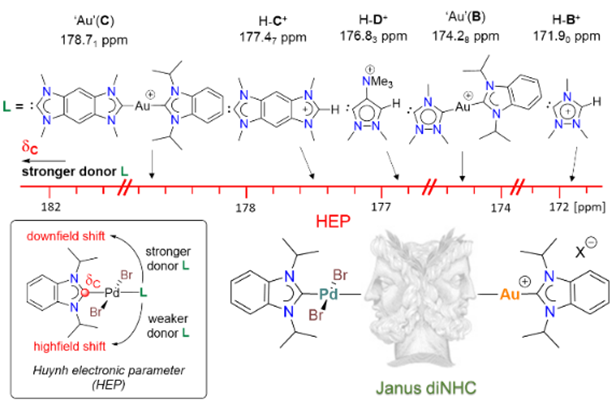
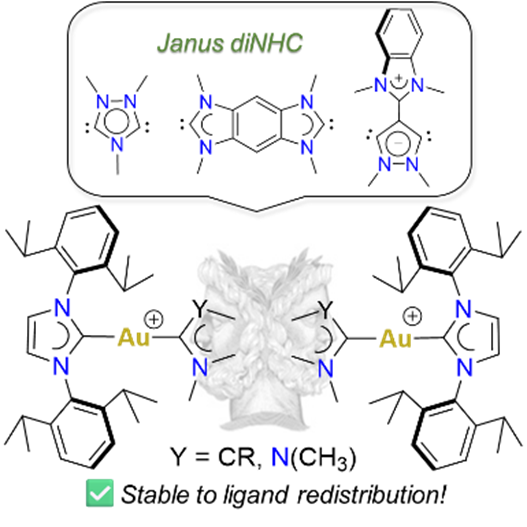














![[triple bond, length as m-dash]](https://www-rsc-org.libproxy1.nus.edu.sg/images/entities/char_e002.gif) N unit, and (iv) methyl acrylate elimination.
N unit, and (iv) methyl acrylate elimination.



























Pd")












, pp 3685–3696")



